15 Jan 2024
As we start the new year, we would like to reflect on the exciting events that kept us busy towards the end of 2023 and announce upcoming MDAnalysis events to look forward to in 2024. We would like to thank the Chan Zuckerberg Initaitve (CZI) and NumFocus for their support in making these events possible.
MDAnalysis UGM (User Group Meeting)
From September 27-29, 2023, 37 MDAnalysis users and developers convened in Lisbon, Portugal for the inaugural MDAnalysis UGM (User Group Meeting). Hosted at the Faculty of Sciences of the University of Lisbon, the event featured presentations and open discussions about using and developing MDAnalysis, followed by a hackathon. Talks covered a broad range of topics including, but not limited to, materials science applications, deep learning and multiscale modeling, drug discovery, and tools for molecular dynamics simulation analysis. Based on attendees’ votes, the following recognitions were awarded to UGM participants:
Best Talk
-
First Place - Namir Oues
-
Second Place (TIE) - Andres Arango, Hannah Baumann
Best Lightning Talk
-
First Place - Hannah Pollak
-
Second Place - Pegerto Fernández
-
Third Place - Hocine El Khaoudi Enyoury
Best Poster
-
First Place (TIE) - Hocine El Khaoudi Enyoury, Dimitris Stamatis
Superstar Pet (According to a pet photo contest on the #mda-pets channel on Discord)
- Pip (owned by Fiona Naughton)
The UGM provided a unique opportunity for MDAnalysis users and developers to interact with other and exchange ideas. Notably, these discussions were key to informing our recently announced roadmap towards MDAnalysis v3.0.
For more details, we invite you to check out the MDAnalysis/UGM2023 GitHub repository, where many of the UGM presentation and hackathon materials may be accessed.


Save the Date for the 2024 MDAnalysis UGM
We are aiming for the UGM to become an annual event and have already begun planning for this year’s meeting. We are therefore excited to announce that the 2024 MDAnalysis UGM will take place August 21-23, 2024 in London, UK!
Additional details about registration and a call for proposals will be communicated soon. In the meantime, save the date and keep an eye out for additional announcements on our blog and UGM website page, as well as on our X (formerly known as Twitter) and LinkedIn accounts.
MDAnalysis Online Training Workshops
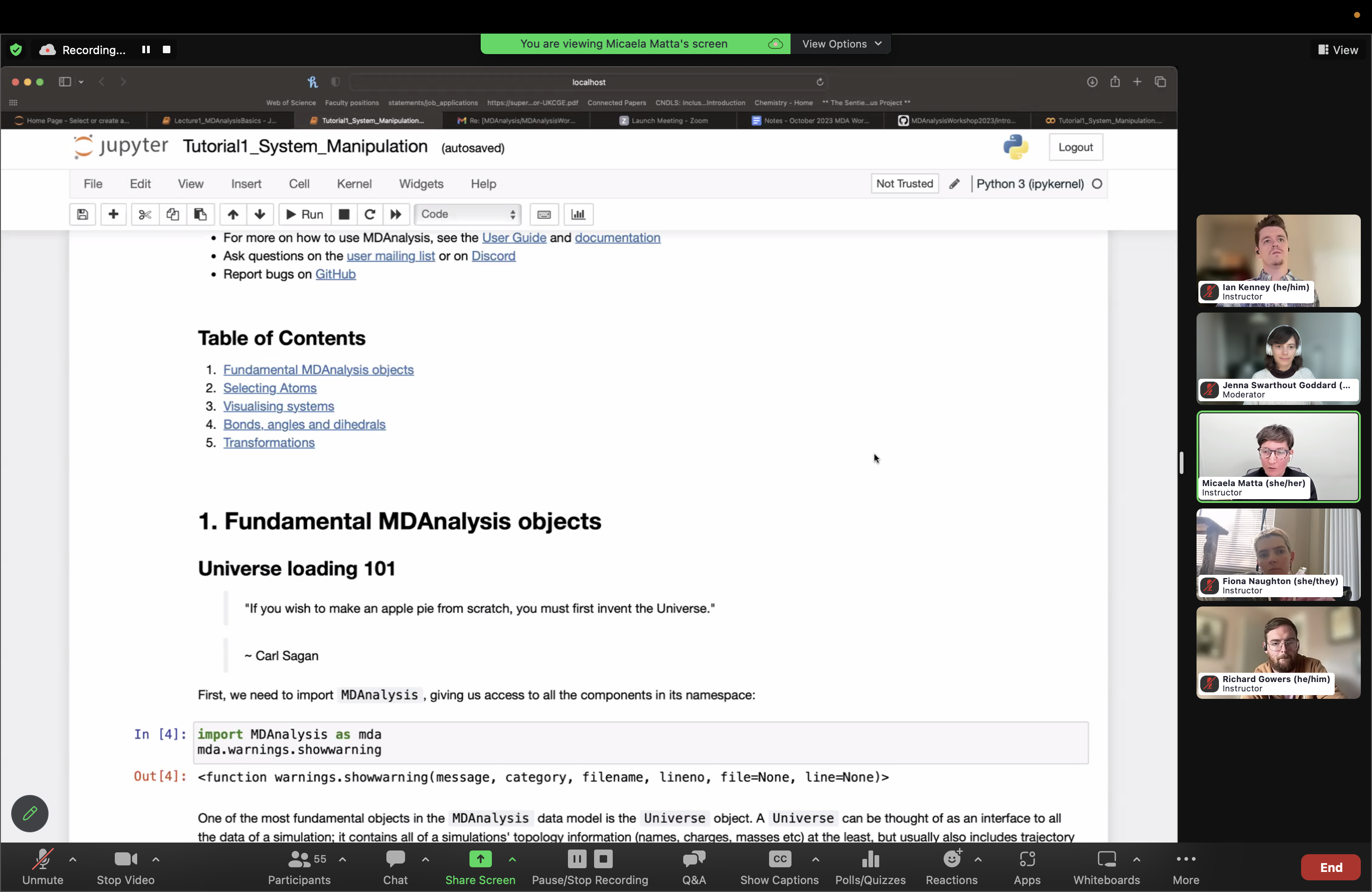
More than 70 people joined us live on October 25, 2023 for a free online workshop, during which 4 instructors (@fiona-naughton, @ianmkenney, @micaela-matta, @richardjgowers) introduced the MDAnalysis package and demonstrated use cases through interactive tutorials; the recording is now available on our YouTube channel. @micaela-matta led the first lecture/tutorial on the Basics of MDAnalysis, and @richardjgowers led a lecture/tutorial on positions, distances, and trajectories; all instructors helped answer participant questions throughout the workshop. Prior to the workshop, @ianmkenney answered participant questions during an optional installation troubleshooting block. All workshop materials and installation instructions are publicly available on the MDAnalysis/MDAnalysisWorkshop2023 GitHub repository.
We will soon be announcing a series of additional workshops taking place in 2024. These workshops are planned to accommodate both beginner and more advanced MDAnalysis users across differing time zones. We are also working on some exciting collaborations to offer workshops on more specialized topics! Stay tuned for updates on our blog, X, and LinkedIn pages.
04 Jan 2024
We are happy to release version 2.7.0 of MDAnalysis!
This is a minor update to the MDAnalysis library.
This release of MDAnalysis is packaged under a
GPLv3+ license.
Additionally all contributions made from commit
44733fc
onwards are made under the
LGPLv2.1+ license.
More details about these license changes can be found in our blog post.
The minimum required NumPy version is 1.22.3.
Supported Python versions: 3.9, 3.10, 3.11, 3.12. Support for version
3.12 has been added in this release.
Supported Operating Systems:
Upgrading to MDAnalysis version 2.7.0
To update with conda from the conda-forge channel run
conda update -c conda-forge mdanalysis
To update from PyPi with pip run
python -m pip install --upgrade MDAnalysis
For more help with installation see the installation instructions in the User Guide.
Make sure you are using a Python version compatible with MDAnalysis
before upgrading (Python >= 3.9).
Notable changes
For a full list of changes, bugfixes and deprecations see the CHANGELOG.
Fixes:
- NoJump now properly handles jumps that occur on the second frame of NPT
trajectories (PR #4258).
- Fixed charge reading from PDBQT files (PR #4283).
- Fixed a case where qcprot.CalcRMSDRotationalMatrix would return a RMSD
of None (PR #4273).
Enhancements:
- Support was added for reading chainID from prmtop AMBER topologies (PR #4007).
- Added support for Python 3.12 (PR #4309, #4300, #4301, #4319, #4325,
#4327, #4329)
- Added support for reading chainID from Autodock PDBQT files (PR #4284), GROMACS
TPR topologies (PR #4281) and amber prmtop topologies (PR #4007).
- Various improvements to the organization and performance of Major and Minor
Pair analyses (PR #3735).
- C distance backend is now exposed via libmdanalysis.pxd (PR #4342).
- Added a GROMOS11 Reader (PR #4294).
Changes:
- Added mda_xdrlib as a core dependency to replace the now deprecated Python
xdrlib code (PR #4271).
- ConverterBase has been moved to MDAnalysis.converters.base (PR #4253).
- networkx is now an optional dependency of MDAnalysis (PR #4331).
- BioPython is now an optional dependency of MDAnalysis (PR #4332).
- Results for WatsonCrickDist nucleic acids analysis are now stored in
analysis.nucleicacids.WatsonCrickDist.results.distances (PR #3735).
Deprecations:
- Importing ConverterBase from MDAnalysis.coordinates.base will not be possible
after MDAnalysis 3.0 (PR #4253).
- Deprecation with intent of removal in MDAnalysis v3.0 of the X3DNA legacy
code (PR #4333).
- Deprecation with intent of removal in MDAnalysis v3.0 of the TRZ reader and
writer (PR #4335).
- Deprecation with intent of removal in MDAnalysis v3.0 of the
MDAnalysis.lib.util.which method (PR #4340).
- The asel argument of the timeseries attribute of Readers is now deprecated
in favour of the atomgroup argument (PR #4343).
- In nucleicacids.WatsonCrickDist, accepting lists of Residue objects was
deprecated in favor of using ResidueGroup: using List[Residue] will be
removed in release 3.0.0; instead use a ResidueGroup (PR #3735).
- In nucleicacids.WatsonCrickDist the result results.pair_distances was
deprecated and will be removed in 3.0.0; use results.distances instead (PR #3735).
Author statistics
This release was the work of 13 contributors, 5 of which are new contributors.
Our new contributors are:
Acknowledgements
MDAnalysis thanks NumFOCUS for its continued support as our fiscal sponsor and
the Chan Zuckerberg Initiative for supporting MDAnalysis under EOSS4 and EOSS5 awards.
— The MDAnalysis Team
21 Dec 2023
Dear MDAnalysis community,
Our Google groups (mailing lists) have been the main way to ask questions, exchange ideas, and to discuss about development.
However, with more modern solutions available we felt a change was needed.
The MDAnalysis Team considered different alternatives and after lengthy discussions we settled on moving the Google groups to GitHub Discussions.
This will have multiple advantages, including proper code formatting and cross-linking between issues and discussions.
GitHub Discussions are already enabled on GitHub and can be used right away: please start using the Discussions forum, instead of the Google groups.
GitHub Discussions are divided into categories, and we will have the following (and more):
We hope to see all of you in the new GitHub Discussions forum!
The mailing lists will remain open for a bit longer to finish off open discussions, after which they will be archived (accessible as read-only).
- Rocco Meli, on behalf of the MDAnalysis Team