Tutorial¶
We will first test that MDAnalysis can perform PSA, using the small test set. We will then look in detail into the radical.pilot script that implements the map-step. Finally, we conclude with the reduce-step and analysis of the data.
Preliminary test¶
Provide topology and trajectory files to the PSA script mdanalysis_psa_partial.py as two lists in a JSON file (see mdanalysis_psa_partial.py for more details). Initially we just want to check that the script can process the data
(mdaenv) $ $RPDIR/mdanalysis_psa_partial.py --inputfile testcase.json -n 5
This means
use the test case
compare the first 5 trajectories against the remaining 5 trajectories
The -n (split) argument is important because we are going to use
it to decompose the full distance matrix into sub-matrices. (If you
just want to do all-vs-all comparisons, use the
mdanalysis_psa.py script.)
You should see output like
Loading paths from JSON file testcase.json
Processing 10 trajectories.
Splitting trajectories in two blocks of length 5 and 5
Calculating D (shape 5 x 5) with 25 entries
----------[ TIMING ]--------------------
load Universes 0.448 s
universe.transfer_to_memory() 0.178 s
PSA distance matrix 1.483 s
saving output 0.001 s
----------------------------------------
total time 2.111 s
----------------------------------------
This indicates that all MDAnalysis parts are working.
Similarly:
(mdaenv) $ $RPDIR/mdanalysis_psa_partial.py --inputfile testcase.json -n 7
should give something like
Loading paths from JSON file testcase.json
Processing 10 trajectories.
Splitting trajectories in two blocks of length 7 and 3
Calculating D (shape 7 x 3) with 21 entries
----------[ TIMING ]--------------------
load Universes 0.422 s
universe.transfer_to_memory() 0.132 s
PSA distance matrix 1.436 s
saving output 0.001 s
----------------------------------------
total time 1.991 s
----------------------------------------
Supercomputer environment¶
We used TACC Stampede to run the calculations on 32 cores but any cluster or even a multi core workstation might be sufficient.
Make sure all env vars are set (especially MongoDB,
RADICAL_PILOT_DBURL) and password-less ssh works.On stampede: Set environment variable
RADICAL_PILOT_PROJECTto your XSEDE allocation:export RADICAL_PILOT_PROJECT=TG-xxxxxx
Activate the mdaenv environment.
You should have the JSON files in the
WORKdirectory.
Copy the two scripts to the WORK directory (at the moment, this is a limitation of the scripts to keep them simple)L
cd WORK
cp $RPDIR/{rp_psa.py,mdanalysis_psa_partial.py} .
Map-step: Radical.pilot script¶
RP script¶
The pilot script is rp/rp_psa.py (see rp_psa.py for details). In the following, important parts of the script are explained.
First, a session is created and a Pilot Manager is initialized:
65 66 67 68 69 70 | session = rp.Session (name=session_name)
c = rp.Context ('ssh')
session.add_context (c)
print "initialize pilot manager ..."
pmgr = rp.PilotManager (session=session)
|
Second, a ComputePilot is described and submitted to the Pilot Manager:
73 74 75 76 77 78 79 80 81 82 | pdesc = rp.ComputePilotDescription ()
pdesc.resource = "xsede.stampede" # xsede.stampede or xsede.comet or ... check docs!
pdesc.runtime = 20 # minutes
pdesc.cores = cores
pdesc.project = PROJECT #Project allocation, pass through env var PROJECT
pdesc.cleanup = False
pdesc.access_schema = 'ssh'
# submit the pilot.
pilot = pmgr.submit_pilots (pdesc)
|
First, a Unit Manager is created for that session and the Pilot is added:
85 86 87 88 | umgr = rp.UnitManager (session=session, scheduler=rp.SCHED_DIRECT_SUBMISSION)
#add pilot to unit manager
umgr.add_pilots(pilot)
|
The script reads the topology and trajectory files from a JSON file:
91 92 | with open(FILELIST) as inp:
topologies, trajectories = json.load(inp)
|
The MDAnalysis script is staged
99 100 101 102 103 104 105 106 107 108 109 110 111 112 | fname_stage = []
# stage all files to the staging area
src_url = 'file://%s/%s' % (os.getcwd(), SHARED_MDA_SCRIPT)
#print src_url
sd_pilot = {'source': src_url,
'target': os.path.join(MY_STAGING_AREA, SHARED_MDA_SCRIPT),
'action': rp.TRANSFER,
}
fname_stage.append(sd_pilot)
# Synchronously stage the data to the pilot
pilot.stage_in(fname_stage)
|
In a loop, a CU is set up for each block matrix — this is the
map step. In particular, the trajectories are partitioned
according to the BLOCK_SIZE (the parameter \(w\)) and all
necessary information is written to a JSON file that will be used as
the input for the mdanalysis_psa_partial.py script:
115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 | cudesc_list = []
for i in range(0, len(trajectories), BLOCK_SIZE):
for j in range(i, len(trajectories), BLOCK_SIZE):
fshared = list()
shared = {'source': os.path.join(MY_STAGING_AREA, SHARED_MDA_SCRIPT),
'target': SHARED_MDA_SCRIPT,
'action': rp.LINK}
fshared.append(shared)
shared = [{'source': 'file://{0}'.format(trajectory),
'target' : basename(trajectory),
'action' : rp.LINK}
for trajectory in trajectories[i:i+BLOCK_SIZE]]
fshared.extend(shared)
if i!=j:
shared = [{'source': 'file://{0}'.format(trajectory),
'target' : basename(trajectory),
'action' : rp.LINK}
for trajectory in trajectories[j:j+BLOCK_SIZE]]
fshared.extend(shared)
# always copy all unique topology files
shared = [{'source': 'file://{0}'.format(topology),
'target' : basename(topology),
'action' : rp.LINK}
for topology in set(topologies)]
fshared.extend(shared)
# block of topology / trajectory pairs
# block[:nsplit] + block[nsplit:]
# The MDA script wants one long list of trajectories and the index nsplit
# that indicates where to split the list to create the two groups of
# trajectories that are compared against each other.
block_top = topologies[i:i+BLOCK_SIZE] + topologies[j:j+BLOCK_SIZE]
block_trj = trajectories[i:i+BLOCK_SIZE] + trajectories[j:j+BLOCK_SIZE]
block = [block_top, block_trj]
nsplit = len(trajectories[i:i+BLOCK_SIZE])
imax = i + len(trajectories[i:i+BLOCK_SIZE])
jmax = j + len(trajectories[j:j+BLOCK_SIZE])
# should remember i, imax and j_jmax because we calculate the
# submatrix D[i:i+di, j:j+dj] in this CU.
block_json = "block-{0}-{1}__{2}-{3}.json".format(
i, imax, j, jmax)
block_matrixfile = 'subdistances_{0}-{1}__{2}-{3}.npy'.format(
i, imax, j, jmax)
manifest.append((block_matrixfile, block_json, (i, imax), (j, jmax)))
# create input file for the cu and add share it
with open(block_json, "w") as out:
json.dump(block, out)
# share input json file
shared = [{'source': 'file://{0}'.format(os.path.realpath(block_json)),
'target' : basename(block_json),
'action' : rp.LINK}
]
fshared.extend(shared)
# define the compute unit, to compute over the trajectory submatrix
# TODO: store the offsets WITH the returned matrix (pkl or arrach archive) instead
# of encoding in filename
cudesc = rp.ComputeUnitDescription()
cudesc.executable = "python"
cudesc.pre_exec = ["module load python; source activate mdaenv"] #Only for Stampede and with our conda env
cudesc.input_staging = fshared
cudesc.output_staging = {'source': block_matrixfile,
'target': block_matrixfile,
'action': rp.TRANSFER}
cudesc.arguments = [SHARED_MDA_SCRIPT, '--nsplit', nsplit,
'--inputfile', block_json,
'--outfile', block_matrixfile, ]
cudesc.cores = 1
cudesc_list.append(cudesc)
|
For the reduce step, all information about the block matrix (filename and indices in the distance matrix) are written to a JSON file (“manifest”):
193 194 | with open(MANIFEST_NAME, "w") as outfile:
json.dump(manifest, outfile)
|
Finally, the CUs are submitted to execute on the compute resources and the script waits until they are all complete:
197 198 199 200 201 202 | # submit, run and wait and...
#print "submit units to unit manager ..."
units = umgr.submit_units(cudesc_list)
#print "wait for units ..."
umgr.wait_units()
|
Launch pilot jobs¶
Launch the pilot job from the login node of stampede (or the cluster where you set up your radical.pilot environment):
python rp_psa.py trajectories.json 20 16 SPIDAL.001
The rp_psa.py radical.pilot script takes as input (see rp_psa.py for details):
the JSON file with the trajectories (trajectories.json)
number of trajectories per block (20)
number of cores to request (16)
session name (arbitrary string, “SPIDAL.001”); note that you need to provide a new session name every time you-rerun the command, e.g. “SPIDAL.002”, “SPIDAL.003”, …
Reduce-step¶
Once the pilot jobs has completed and all block matrices have been computed we combine all data (reduce) into the distance matrix \(D_{ij}\) and analyze it:
psa_reduce.py -t trajectories.json -m manifest.json \
-p psa_plot.pdf -o distance_matrix.npy
Combine block matrices¶
The manifest.json file contains all information that is needed to
re-assemble the distance matrix: for each output file it also stores
the indices of the sub-matrix in the distance matrix and so the full
distance matrix can be built with
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | def combine(traj, manifest):
trj = json.load(open(traj))
N = len(trj[0])
D = np.zeros((N, N))
mf = json.load(open(manifest))
missing = []
for subD, _, (i0, i1), (j0, j1) in mf:
try:
D[i0:i1, j0:j1] = np.load(subD)
D[j0:j1, i0:i1] = D[i0:i1, j0:j1].T
except IOError:
missing.append(subD)
if missing:
print("WARNING: Missing data: output files\n{}\n"
"could not be found".format(", ".join(missing)))
return D
|
The matrix is written to a numpy file distance_matrix.npy (and can
be loaded with numpy.load()).
Analysis¶
The distance matrix can be clustered to reveal patterns in the
trajectories. Here we use hierarchical clustering with Ward’s linkage
criterion as implemented in scipy.cluster.hierarchy
[Seyler2015]. The clustering and plotting code was taken from the
cluster() and
plot() methods of
PSAnalysis. The plotting code is
fairly complicated but the clustering is straightforward:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 | def cluster(distArray, method='ward', count_sort=False,
distance_sort=False, no_plot=False, no_labels=True,
color_threshold=4):
"""Cluster trajectories and optionally plot the dendrogram.
:Arguments:
*method*
string, name of linkage criterion for clustering [``'ward'``]
*no_plot*
boolean, if ``True``, do not render the dendrogram [``False``]
*no_labels*
boolean, if ``True`` then do not label dendrogram [``True``]
*color_threshold*
For brevity, let t be the color_threshold. Colors all the
descendent links below a cluster node k the same color if k is
the first node below the cut threshold t. All links connecting
nodes with distances greater than or equal to the threshold are
colored blue. If t is less than or equal to zero, all nodes are
colored blue. If color_threshold is None or ‘default’,
corresponding with MATLAB(TM) behavior, the threshold is set to
0.7*max(Z[:,2]). [``4``]]
:Returns:
list of indices representing the row-wise order of the objects
after clustering
"""
matplotlib.rcParams['lines.linewidth'] = 0.5
Z = scipy.cluster.hierarchy.linkage(distArray, method=method)
dgram = scipy.cluster.hierarchy.dendrogram(
Z, no_labels=no_labels, orientation='left',
count_sort=count_sort, distance_sort=distance_sort,
no_plot=no_plot, color_threshold=color_threshold)
return Z, dgram
|
In the resulting plot we indicate FRODA trajectories with a heavy double-bar and DIMS trajectories with a thin single bar.
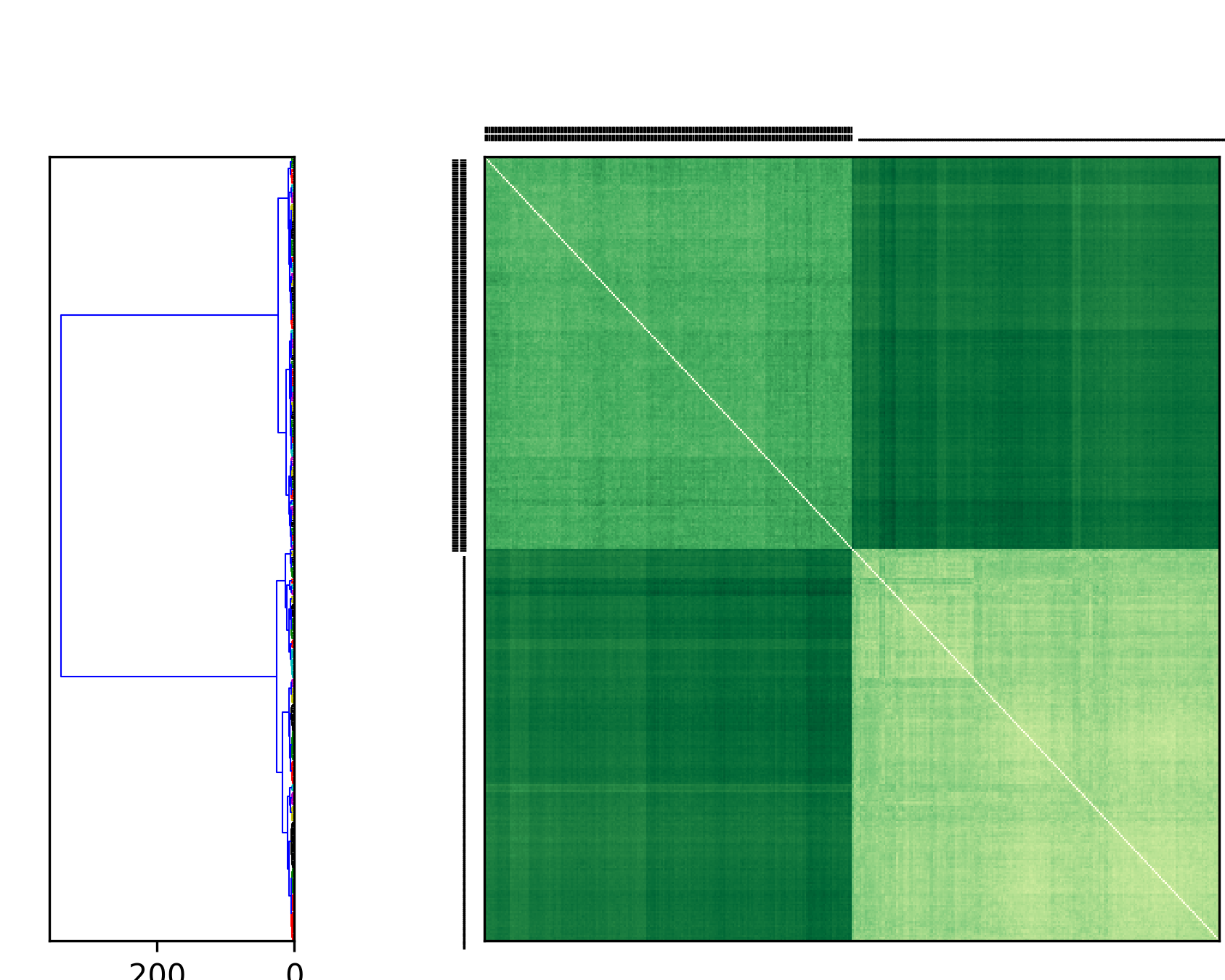
PSA of AdK transitions. Transitions of AdK from the closed to the open conformation were generated with the DIMS MD and the FRODA method. Path similarity analysis (PSA) was performed by hierarchically clustering the matrix of Fréchet distances between all trajectories, using Ward’s linkage criterion.¶
Symbol |
Method |
|---|---|
|| |
FRODA |
| |
DIMS |
Figure PSA of AdK transitions shows clearly that DIMS and FRODA transitions are different from each other. Each method produces transitions that are characteristic of the method itself. In a comparison of many different transition path sampling methods it was also found that this pattern generally holds, which indicates that there is likely no “best” method yet [Seyler2015].
