GSoC Report: From RDKit to the Universe and back 🚀
29 Aug 2020With the end of the summer comes the end of my awesome Google Summer of Code adventure with the MDAnalysis team. It’s also the occasion for me to report on the code I’ve implemented, the things I’ve learned and the challenges I’ve faced.
Summary of the project
The goal of my project, From RDKit to the Universe and back, was to provide interoperability between MDAnalysis and RDKit. i.e. to be able to:
- Read an RDKit Mol and create an MDAnalysis Universe with it,
- Convert an MDAnalysis AtomGroup to an RDKit Mol,
- Leverage RDKit’s functionalities directly from MDAnalysis (descriptors, fingerprints, aromaticity perception…etc.)
With this in mind, the project was easily cut down in 2 main deliverables (the RDKit Reader/Parser and the Converter) and a few smaller ones for each “wrapper” functionality.
The motivation behind the interoperability project was to be able to benefit from all the features that are available in RDKit and MDAnalysis with as little hassle as possible.
For example, before this project if you wanted to compute molecular descriptors for a ligand in an MD trajectory, you would have to write a separate PDB file for each frame of the trajectory and then read each file through RDKit. With the work that I’ve done, you can now convert your MD trajectory to an RDKit molecule and compute descriptors from there, or you can directly use the descriptor wrapper on an MDAnalysis AtomGroup (see below).
It was also the occasion for me to increase my visibility in the community by working on an open-source software development project, and to learn how to write better code. After my PhD I’d like to develop software for computational chemistry and this Google-sponsored event will hopefully help me in that regard.
Contributions
Merged PRs
-
Converting an RDKit molecule to MDAnalysis: #2707
This was the first part of the project and it helped me get acquainted with MDAnalysis as I had never used it before GSoC. The goal here was to be able to “parse” an RDKit molecule and build an MDAnalysis Universe from it.
It was also the occasion for me to start interfacing MDAnalysis with RDKit as I got to implement a new classmethod for the Universe, theUniverse.from_smilesmethod which allows us to build a Universe from a SMILES string, but also to add atom selection based on aromaticity.
At the end of this first PR, I was familiar with the different core objects that compose the Universe as well as writing tests with pytest and documentation with sphinx. -
Converting an MDAnalysis universe to RDKit: #2775
The second goal of the project was to be able to convert an MDAnalysis AtomGroup to an RDKit molecule, allowing users to analyse MD trajectories in RDKit. While this should be trivial once we know how to do the opposite operation, it was actually a real challenge to get a molecule with the correct bond order and charges out of it.
As you may know, most MD topology file formats don’t keep track of bond orders and formal charges so we have to find a way to infer this information from what we have. In our case, we require all hydrogen atoms to be explicit in the topology file, as well as elements and bonds (although these two can be guessed). Then the bond orders and formal charges are inferred based on atomic valencies and the number of unpaired electrons, followed by a standardization step of functional groups and conjugated systems. This last step is needed because the algorithm implemented to guess bond orders and charges is dependent on the order in which atoms are read.
Let’s take azathioprine as an example to visualize the different steps of the RDKitConverter:
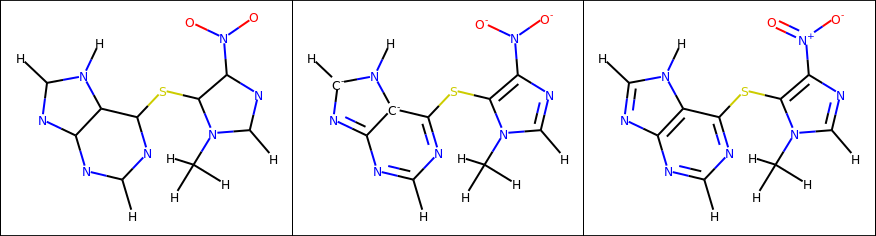 On the left is what you would get from a typical topology file: elements and bonds between atoms, but nothing more. In the middle, we’ve inferred bond orders and charges but because of the order in which atoms were read, two carbon atoms that were supposed to be part of the conjugated system end up negatively charged, and the nitro group isn’t represented in its usual form. On the right, we’ve corrected the purine ring and standardized the nitro group to obtain the final molecule.
On the left is what you would get from a typical topology file: elements and bonds between atoms, but nothing more. In the middle, we’ve inferred bond orders and charges but because of the order in which atoms were read, two carbon atoms that were supposed to be part of the conjugated system end up negatively charged, and the nitro group isn’t represented in its usual form. On the right, we’ve corrected the purine ring and standardized the nitro group to obtain the final molecule.
This took more time than originally planned in the project timeline but was well worth it. -
SMARTS selection: #2883
SMARTS is an extension of the SMILES language that is used for substructure searching. Being able to select atoms based on SMARTS queries, and combine these selections with those already available in MDAnalysis might be one of the key features that will come out of this project.
In progress
-
Wrap RDKit drawing code for AtomGroups: #2900
This PR allows us to draw images (SVG, PNG, and GIF) of AtomGroups using RDKit. It also adds rich displays to AtomGroups in notebooks (a.k.a.__repr__methods). Before working on this, I thought the only way to use alternative representations for Python objects was to define a_repr_*_method for your class, where * is a MIME type such as png, html…etc. There is actually a second way, where you tell IPython directly how it’s supposed to represent an object. This allows funky representations of any object, even python built-in types, i.e.intas roman numerals and so on. I also wrote my first metaclass here, to register different “viewer” classes when more become available in the future. It’s also not straightforward to write tests for the images as different versions of RDKit or other packages will lead to slightly different outputs. More discussion, code review, and tests are needed before this is ready. -
Wrap RDKit descriptors and fingerprints: #2912
This PR adds new kinds of analysis that are typically performed on small molecules in the chemoinformatics field. Fingerprints are mostly used for calculating similarity metrics between molecules and a reference. Molecular descriptors could be used to describe all the sampled conformations of a ligand in a binding pocket during a simulation, and given as input to a machine-learning model for clustering, scoring binding poses…etc.
This is currently missing more discussion, documentation, and a few tests. -
Change the Converters API: #2882
While developing the RDKitConverter, some interesting points were made about the current API used to convert AtomGroups, i.e.u.atoms.convert_to("RDKIT"). This method is case sensitive, it doesn’t allow to pass arguments to the converter class, and it requires users to read the documentation to know which converters are available. This PR corrects the two first points and adds the possibility to either use the previous syntax or tab-completion to find the available converters i.e.u.atoms.convert_to.rdkit().
This was inspired by pandasdf.plot(kind="scatter", ...)which is also accessible asdf.plot.scatter(...).
Since this is an API change, more discussion with core developers is needed for now.
Left to do
- Guessers for aromaticity and Gasteiger charges through RDKit
- Tutorial on the reader/converter and wrapped RDKit functionalities in the UserGuide. This will make it easier for users to know about the features I implemented and how to use them properly, rather than searching for every single feature in the documentation.
- Documentation on the RDKit format in the UserGuide: #69
Demo
Full circle
Here are all the possible conversions between RDKit and MDAnalysis:
import MDAnalysis as mda
from rdkit import Chem
# new feature
u1 = mda.Universe.from_smiles("CCO")
# new feature
mol1 = u1.atoms.convert_to("RDKIT")
# new feature
u2 = mda.Universe(mol1)
# before this project
u2.atoms.write("mol.pdb")
mol2 = Chem.MolFromPDBFile("mol.pdb")
Atom selections
There are two new selections available in MDAnalysis: aromatic for aromatic atoms, and smarts for the selection of atoms based on SMARTS queries. Let’s try them on this molecule:
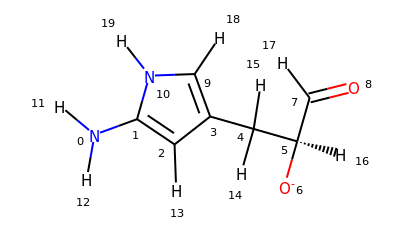
>>> u = mda.Universe.from_smiles("Nc1cc(C[C@H]([O-])C=O)c[nH]1")
>>> u
<Universe with 20 atoms>
>>> u.select_atoms("aromatic")
<AtomGroup with 5 atoms>
# same as above
>>> u.select_atoms("smarts a")
<AtomGroup with 5 atoms>
# 4 aromatic carbon atoms
>>> u.select_atoms("smarts c").indices
array([1, 2, 3, 9])
# carbon atoms in a ring (not necessarily aromatic)
>>> u.select_atoms("smarts [#6;R]").indices
array([1, 2, 3, 9])
# 1 aromatic nitrogen
>>> u.select_atoms("smarts n").indices
array([10])
# not hydrogen and not in a ring but connected to a ring
>>> u.select_atoms("smarts [$([!R][R])] and not type H").indices
array([0, 4])
Descriptors calculation
Soon, you will be able to compute descriptors directly from an AtomGroup, by either passing the name of the descriptor in RDKit, or by passing your own function that takes an RDKit molecule as argument. Here’s an example of what the current version looks like:
>>> from MDAnalysis.analysis.RDKit import RDKitDescriptors
>>> u = mda.Universe.from_smiles("CCO", numConfs=3)
>>> def num_atoms(mol):
... return mol.GetNumAtoms()
>>> desc = RDKitDescriptors(u.atoms, "MolWt", "RadiusOfGyration",
... num_atoms).run()
>>> desc.results
array([[46.06900000000002, 1.161278342193013, 9],
[46.06900000000002, 1.175492972121405, 9],
[46.06900000000002, 1.173230936577319, 9]],
dtype=object)
Fingerprint calculation
You will also be able to obtain fingerprints:
>>> from MDAnalysis.analysis.RDKit import get_fingerprint
>>> fp = get_fingerprint(u.atoms, "AtomPair", hashed=True, nBits=1024)
>>> fp.GetNonzeroElements()
{36: 1,
106: 1,
297: 1,
569: 3,
619: 1,
624: 8,
634: 2,
699: 1,
745: 5,
819: 4,
938: 6,
945: 3}
Drawing with RDKit
Finally, you will be able to display small AtomGroups as images in notebooks:
>>> from MDAnalysis.visualization.RDKit import RDKitDrawer
>>> from nglview.datafiles import PDB, XTC
>>> u = mda.Universe(PDB, XTC)
>>> elements = mda.topology.guessers.guess_types(u.atoms.names)
>>> u.add_TopologyAttr('elements', elements)
>>> u.atoms
<AtomGroup with 5547 atoms>
>>> ag = u.select_atoms("resname LRT")
>>> ag

You can also export any AtomGroup to a PNG or SVG, and even to a GIF for trajectories.
Conclusion
This project taught me a lot of things on software development and the Google Summer of Code experience has been incredible and valuable to me. All of this wouldn’t have been possible without the help of many people, including my amazing mentors (@IAlibay, @fiona-naughton and @richardjgowers), but also the rest of the MDAnalysis team as a lot of them got involved and gave me great feedback, so thank you to all of them!
— @cbouy